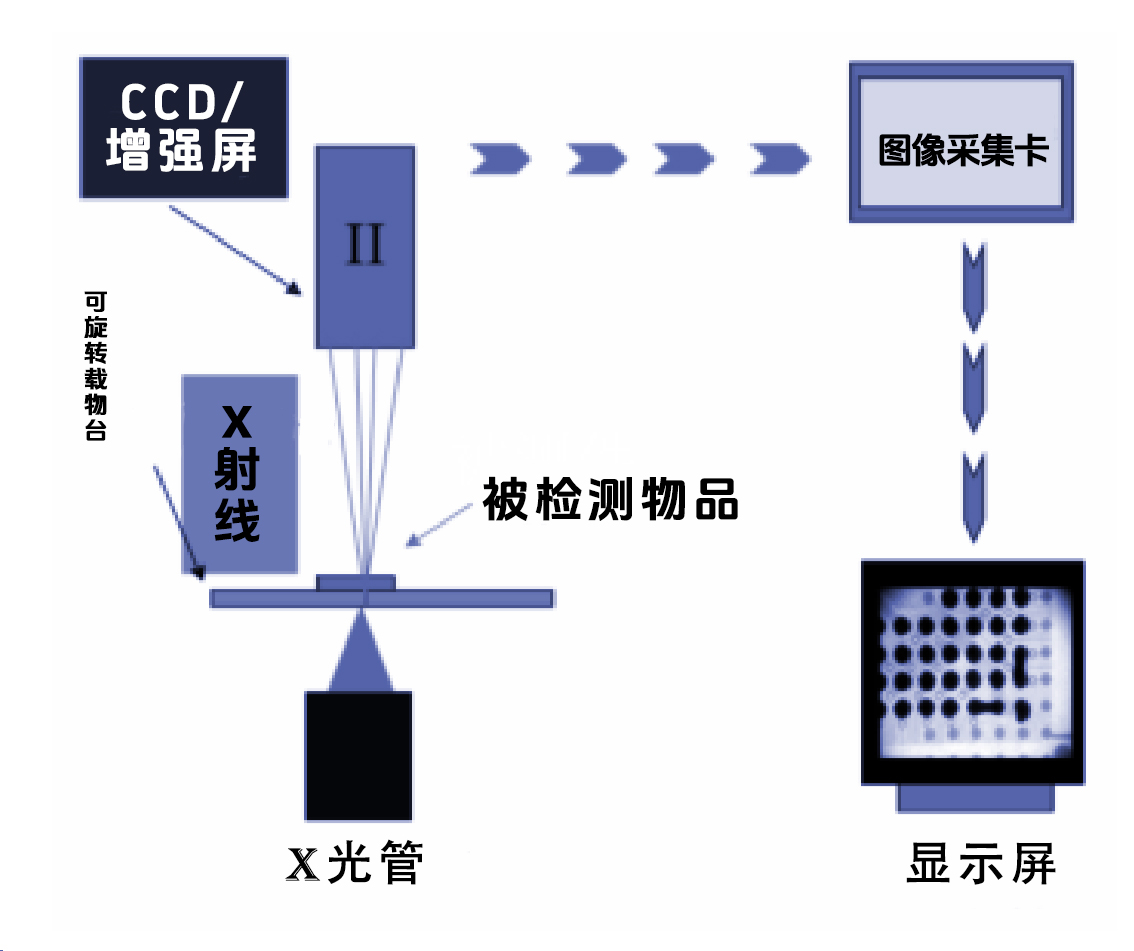
2022年5月26日,欧盟体外诊断医疗器械法规(EU)2017/746(IVDR)正式实施。
背景
2017年4月5日,欧洲议会和理事会通过有关体外诊断医疗器械法规(EU)2017/746(简称“IVDR”),同时废除指令98/79/EC(简称“IVDD”)和委员会决议2010/227/EU。2017年5月5日欧盟官方公报OJ L 117正式发布IVDR法规,同年5月26日生效,过渡期为五年,今天是法规正式实施的日期。
法规主要内容
IVDR法规共十章113条,15个附录。
|
章节(条款) |
标题 |
|
第I章(条款1-4) |
范围和定义 |
|
第II章(条款5-21) |
器械上市和投入使用;制造商、进口商和运营商等的责任;CE认证;欧盟成员内自由移动 |
|
第III章(条款22-30) |
器械的标识和可追溯性;器械和运营商的注册;安全和临床性能总结;欧洲医疗器械数据库(“Eudamed”) |
|
第IV章(条款31-46) |
CE认证公告机构(Notified Bodies) |
|
第V章(条款47-55) |
器械分类和合格评定 |
|
第VI章(条款56-77) |
临床实证、性能评价和性能研究 |
|
第VII章(条款78-95) |
上市后监督、警戒和市场监督 |
|
第VIII章(条款96-101) |
欧盟成员国、医疗器械协调小组、欧盟参考实验室和器械注册机构之间的合作 |
|
第IX章(条款102-106) |
保密性、数据保护、资金和处罚 |
|
第X章(条款107-113) |
最终条款 |
|
附件I |
通用安全和性能要求 |
|
附件II |
技术文件 |
|
附件III |
上市后监督的技术文件 |
|
附件IV |
欧盟符合性声明 |
|
附件V |
CE认证标记 |
|
附件VI |
根据第26(3)条和第28条,注册器械和运营商需要提交的信息;根据第25条和第26条,与UDI-DI一起提供给UDI(医疗器械唯一标识)数据库的核心数据元素,以及UDI系统 |
|
附件VII |
公告机构应满足的要求 |
|
附件VIII |
分类规则 |
|
附件IX |
基于质量管理体系和技术文件评估的合格评定 |
|
附件X |
基于型式检验的合格评定 |
|
附件XI |
基于生产质量保证的合格评定 |
|
附件XII |
由公告机构颁发的证书 |
|
附件XIII |
性能评估、性能研究和上市后性能跟踪 |
|
附件XIV |
干预性临床性能研究和某些其他性能研究 |
|
附件XV |
旧指令98/79/EC和本法规条款的对比表 |
新法规的主要变化
1. 扩大产品范围。
2. 根据风险对器械进行重新分类。
3. 更严格的技术文档要求。
4. 确定“负责监管合规的人员”。
5. 增加公告机构的参与,对公告机构进行更严格的审查。
6. 实施唯一设备标识(UDI),以实现更好的可追溯性。
特别提醒
新法规的变化意味着,欧盟对进入欧洲市场的医疗器械将实施更严格的限制,对行业从业者也提出了更高的要求。随着新法规的正式实施,提醒相关行业及时了解新规要求并做好CE认证的更新,避免遭受不必要的损失。
END