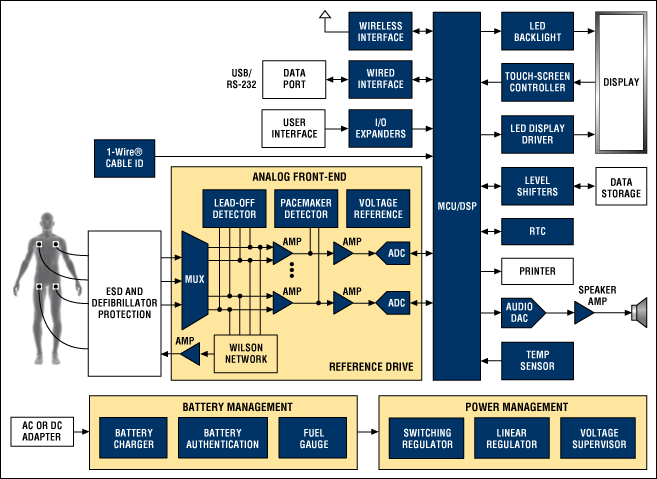
一、FDA最新医疗器械相关法规动态
美国FDA正持续推进监管现代化。最值得关注的变革是:质量体系法规QSR 820正式与ISO 13485:2016协调,于2024年2月2日发布最终规则,形成新的质量管理体系法规QMSR,并于2026年2月2日全面生效。此外,对软件、人工智能医疗器械、网络安全等方面的指南不断更新,体现了对数字健康的强化监管。
二、美国医疗器械法规框架核心
- FDA医疗器械的分类及监管方式
- 基于风险,医疗器械分为I、II、III类,风险逐级递增。
- 监管方式:I类(一般控制,大部分豁免510k)→ II类(特殊控制+510k)→ III类(上市前批准PMA)。
- 医疗器械产品分类确定及实例
- 确定方法:通过产品代码、分类法规、数据库比对,或提交513(g)分类申请。
- 实例:血糖仪通常为II类(产品代码NGH),需510(k);人工心脏瓣膜为III类,需PMA。
三、510(k)申报核心要点
- 510(k)资料主要内容及要求
- 必须包括:实质等同声明、对比器械论证、适用范围、技术性能数据、生物相容性、软件验证、灭菌验证等。
- 关键文件:510(k)摘要/声明是公开信息,需精心撰写以清晰呈现等同逻辑。
- 510(k)递交流程
- 步骤:确定分类与对比器械→准备测试与数据→撰写提交材料→通过电子提交网关CESG提交→缴纳用户费→FDA进行受理审查→实质性互动审查→最终决定。
- MDUFA时间线:目标在100个自然日内完成(受理审查15天,实质审查至决定90天)。
- 510(k)决定与异常处理
- 决定类型:实质等同(SE,获准上市)、非实质等同(NSE,拒绝)、或要求补充信息(AI)。
- 异常处理:
- RTA暂缓:资料不全,不予受理。
- NSE后:可申请重新分类(De Novo)或重新提交。
- 对决定有异议:可在30天内申请复审。
- 510(k)相关问题剖析
- 常见拒因:对比器械选择不当、数据不足以支持等同性、忽视新的安全有效性问题。
- 策略建议:早期与FDA进行预提交会议,可显著降低风险、明确方向。
QSR 820/QMSR通用部分——构建持续合规的基石
一、ISO 13485与21 CFR 820/QMSR的核心差异
- 法律效力:QMSR是美国联邦法规,具有法律强制性;ISO 13485是国际标准,通常为认证/市场准入所需。
- 监管重点:QMSR更强调设计控制、CAPA、供应商控制以及与FDA的报告义务(如不良事件、召回)。
- QMSR新框架:在融合ISO 13485结构的基础上,保留了FDA独特的监管要求,如医疗器械报告MDR。
二、QSR 820向QMSR的主要修订内容
- 全面采纳ISO 13485:2016的条款和术语作为法规基础。
- 明确“基于风险的方法”贯穿于质量管理体系全过程。
- 对“临床评估”、“上市后监督”等提出了更明确的要求。
- 保留了FDA特有的报告和检查要求。
三、FDA工厂审核依据及可能结果
- 审核依据:QMSR法规是核心依据,审核将验证体系运行的符合性与有效性。
- 可能结果:
- 无行动指示:轻微观察项,书面回应即可。
- 483表:现场检查观察项,需在15个工作日内书面回复。
- 警告信:严重违规,需立即整改,否则可能面临禁令、扣押、起诉。
- 进口警报:产品在入境时将被自动扣留。
四、QMSR控制子系统重点与应对
- 管理控制子系统:强调管理层责任、质量方针、内审和管理评审。应对:确保最高管理者深度参与,资源配备充足。
- 设计控制子系统:从用户需求到设计转换的全过程控制。应对:建立完整的DHF,确保设计变更受控。
- 生产和流程控制子系统:涵盖环境控制、设备校验、物料管理、生产过程与最终检验。应对:建立稳健的DHR,实现全过程可追溯。
- 纠正预防措施子系统:不仅是处理投诉,更是从数据分析中前瞻性预防。应对:建立从数据来源到措施有效性验证的闭环。
- 文件/记录控制子系统:确保所有质量活动有据可查、版本受控。应对:电子化质量管理系统是高效管理的关键。
五、如何有效应对FDA工厂审核
- 审核问题案例分析:
- 典型缺陷:CAPA流于形式、设计变更未经验证、供应商评估不足、培训记录缺失。
- 根源:体系与实际运行“两张皮”,未能贯彻基于风险的思想。
- FDA工厂审核应对策略:
- 日常准备:模拟审核、定期内审、管理层评审。
- 现场应对:由经验丰富的负责人主导,诚实、直接、及时地提供记录。
- 事后整改:对483表或警告信,必须进行根本原因分析,制定包含系统性预防措施的整改计划,并证明其有效性。
驾驭美国双轨合规,需要系统化的专业伙伴
从复杂的510(k)技术论证,到严密的QMSR体系构建与维护,再到应对FDA现场审核,每一环节都环环相扣,充满专业挑战。企业单打独斗,往往面临信息滞后、理解偏差、资源分散的困境,极易导致上市延迟、审核失败甚至监管处罚。
SPICA角宿咨询,作为您深耕全球医疗器械合规的专业战略伙伴,提供“上市前+上市后”的一站式、系统化解决方案:
- 510(k)高效通关:我们从产品分类策略、对比器械优选、测试方案设计到全套技术文件撰写与提交,提供全流程项目管理与代理服务,最大化您的首次通过率,精准控制时间线。
- QMSR体系整合与提升:我们不仅帮助您建立或优化符合QMSR要求的质量管理体系,更能确保其与ISO 13485无缝整合,杜绝“两张皮”,实现从合规到卓越运营的跨越。
- FDA审核全程护航:我们提供从模拟审核、差距分析、人员培训,到现场陪检支持、483表/警告信应对整改的全面服务,助您自信、从容地通过FDA检验。
- 持续合规支持:我们提供法规联络人、定期法规更新解读、上市后监督系统建立等持续服务,确保您的合规状态与时俱进,业务永续经营。
美国市场机遇与挑战并存。立即联系SPICA角宿咨询,让我们以体系化的专业能力,为您扫清合规障碍,筑牢质量基石,稳健开拓全球高端市场。